I Have recently discovered this technique, which can allow genomic sequencing of single virions. Usually the process requires cultivable virus-host systems, given the fact that even when using environmental metagenomics we are still dependent, for the most part, on reference genomes that have been sequenced and uploaded to gene data banks via cultivable viruses.
Some of the currently used techniques are shotgun sequencing (involves randomly breaking up DNA sequences into lots of small pieces and then reassembling the sequence by looking for regions of overlap), tiled amplicon PCR (which allows sequencing samples with a low viral genome copy number for a given known target genome), Multiple displacement amplification (MDA, which does not employ sequence-specific primers but amplifies all DNA), etc.
In relation to the no-virus people, this is their main objection, the process by which viruses are cultured, enriched and sequenced. In their opinion, the material has not been purified to the level of pure virions, and therefore the sequences obtained cannot be determined to belong to any virus. I am not going to discuss further their objections or the methods that are currently used, their accuracy and error rate, or anything else for that matter here. I will only focus on this relatively new approach.
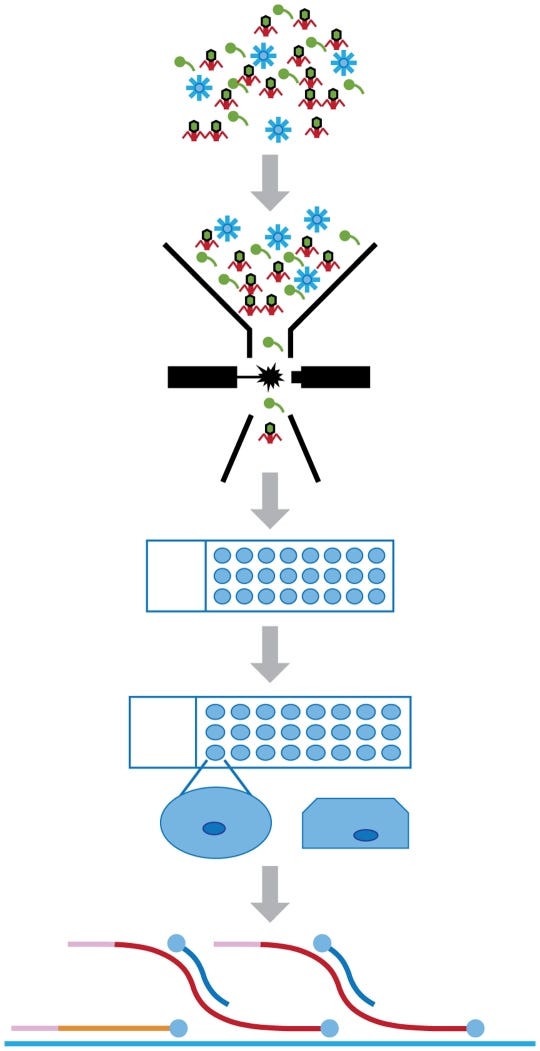
Here we introduce an approach for isolating and characterizing the genomes of viruses called “Single Virus Genomics” (SVG) (Figure 1). The benefits of SVG will be far-reaching, enabling novel virus discovery in a variety of clinical and environmental settings, altering our understanding of virus evolution, adaptation and ecology and facilitating the interpretation of viral genomic and metagenomic data by providing suitable reference genomes.
amplification of genomic material via MDA was successful in 92% of virus-containing droplets.
With the exception of the first 5 bp, the complete genome of lambda was recovered. Lacking the first 5 bp is likely due to a reported artifact of MDA reactions where the ends of linear DNA segments are underrepresented.
De novo assembly was performed with the GS De Novo Assembler Software (i.e., Newbler, 454 Life Sciences) to assess the utility of these methods for use on unknown SVGs.
This method of bioinformatic normalization of the data resulted in larger contigs coupled with almost complete coverage of the genome (>99%). The utility of SVG approaches for the study of uncultivated viruses will ultimately depend on the success of de novo assembly due to the lack of suitable reference genomes.
The Single Virus Genomics approach described here enabled, for the first time, isolation and whole genome sequencing of an individual virus; a significant technical achievement that has the potential to alter the course of virological research.
If you carefully read the paper, they encountered several difficulties and their results are not optimal, nonetheless they are quite impressive, being the first time this has been done. As they indicate themselves “To date, complete coverage of bacterial genomes has not been reported, suggesting that single cell genomics projects suffer from similar obstacles.”
Single-virus genomics reveals hidden cosmopolitan and abundant viruses, 2017.
Here we apply single-virus genomics (SVGs) to assess whether portions of marine viral communities are missed by current techniques. The majority of the here-identified 44 viral single-amplified genomes (vSAGs) are more abundant in global ocean virome data sets than published metagenome-assembled viral genomes or isolates.
using FAVS [fluorescence-activated virus sorting] in combination with confocal fluorescence microscopy, we demonstrated the suitability of the used flow cytometer sorter to separate individual viral particles from a culture isolate.
To explain why our discovered abundant viruses have been overlooked by metagenomic assembly… In our study, the abundant surface vSAGs populations showed high accumulated microdiversity in the diversity curves against the viromes from the corresponding sampling sites. However, in comparison to viromics for those abundant viral population from Tara data set, a contrasting population structure lacking microdiversity was observed. Furthermore, the frequency of single-nucleotide polymorphisms (SNPs) in the vSAG 37-F6 species population was ~10 to 250-fold higher.
Therefore, we hypothesized that population microdiversity would hinder genome reconstruction by metagenomic assembly, which may explain why metagenomics have so far failed to recover some very abundant marine viruses.
This last part is extremely relevant in relation to the background signal for the SARS-like coronavirus quasispecies and the completely underrepresented variations that most likely were already present before 2019, like the Omicron ancestry, for instance, which is missing.
Benchmarking of Single-Virus Genomics: a new tool for uncovering the virosphere, 2020.
I only found access to the Manuscript submitted to Environmental Microbiology, here:
Single-virus genomics (SVG) have recently enabled the discovery of widespread and abundant uncultured viruses in nature by sequencing one virus at a time directly collected from the environment.
First, accurate flow cytometric detection and sorting of viruses fluorescently stained with SYBR dye is critical for the overall success of SVG. When working with biological samples expected to contain high levels of cellular debris or extracellular DNA (e.g., wastewater, stool samples or a bacterial culture such as the pure T4 culture used herein), we strongly recommend the implementation of a DNase digestion step.
Vesicles that have a similar virus size are also abundant in environmental samples (Biller et al., 2014) and sometimes vesicles contain sufficient DNA to be visible with SYBR dyes and thus could be confounded with viruses. However, it has been demonstrated that only a very small proportion of vesicles in environmental samples (<0.01–1%) package enough DNA material to be visible by SYBR staining (Biller et al., 2017).
As before, without entering in too much detail, the researchers also found several difficulties and their results were not optimal. Another important factor to consider is the type of viruses used, mostly phage and marine viruses. It would be very much interesting, once this methodology starts to solve the main hindrances that it has encountered, to see how many errors have we committed in the characterization of different virus species via cultivable virus-host systems. I am sure many controversies may arise.
FINAL NOTE:
As I already mentioned, this new technique does not solve the problem of viruses as disease-causing agents, which in my opinion is very misleading and simplistic. Probably the no-virus people should focus exclusively on this last part. They may increase their credibility rate considerably that way. In order to say that viruses do not exist at all you must first discredit the whole of Molecular Biology, not a very smart approach in my opinion.
Thanks for reading Agus_Z’s Newsletter! Subscribe for free to receive new posts and support my work.